The Bioinformatics Core will enable Systems-Guided Forward Genetics by applying multiple types of computational analyses to three problems: (i) identifying genes likely to regulate critical immune functions so that mutations mapped to these genes can be enriched in a pedigree or selected for in-depth phenotypic analysis; (ii) identifying the molecular basis of an immune phenotype arising from a given point mutation, and (iii) predicting immune pathways that may be dysregulated in immune cells bearing a given mutation. The Bioinformatics Core will support the individual Projects as well as the overall goals of the program through integrated computational analysis of all large-scale datasets generated by the program.
Aim 1
Analyze and integrate transcriptomic, genomic/epigenomic, proteomic, and multiparameter flow cytometry data in support of the projects within the program.
The core will develop and continually update ranked lists of candidate and immune regulatory genes. Computational methods will be used to identify genes, gene signatures, splice forms, or epigenetic features that are predictive of complex immune phenotypes arising from genetic perturbations or pathogenic infection. The core will integrate large-scale datasets from immune cells and relevant external datasets to identify transcriptional regulatory networks and protein interaction networks that are perturbed or dysregulated in immune cells bearing mutations in genes of interest or mutations with immune phenotypes.
Aim 2
Coordinate dissemination of Project data and information about Project resources such as reagents, computational tools, and mutant mouse strains to the scientific community.
The core will maintain and update the consortium website as the program progresses, as well as implement the resource sharing plans.
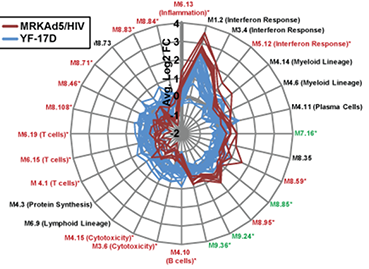
Gene modules and network visualization simplify complex datasets and facilitate hypothesis formulation.
Radar plot comparing innate immune responses of humans to two different viral vaccines, the Merck candidate HIV vaccine (red lines) and a yellow fever virus vaccine. Each spoke is a gene module (see accompanying text) and the radius represents the module’s average absolute fold-change in PBMCs post-vaccination.
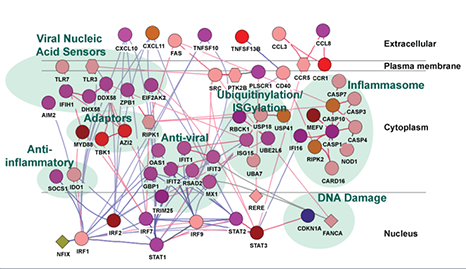
Interaction network representation of genes induced in PBMCs by HIV vaccination in vivo and in vitro, demonstrating vaccine-induced coordinate regulation of major innate immune pathways (highlighted in blue). Nodes (genes) are colored according to module membership, and node shapes indicate type of differential expression (ovals = gene level, diamonds = exon level). Edges indicate interactions (red = protein-protein, blue = protein-DNA).