Project Two
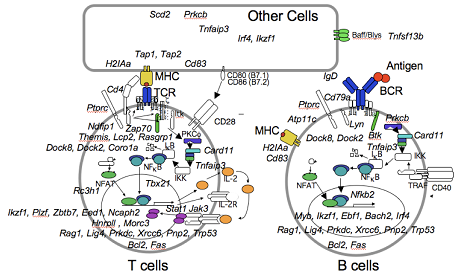
Control systems governing short vs long-term humoral & cellular immunity
Adaptive immunity to pathogens hinges upon separate processes for short- and long-term increases in the activity of antigen-specific B and T lymphocytes. Short-term multiplication and differentiation of plasma cells and cytotoxic T cells is critical for resolving an existing infection, but the strength of the immediate response needs to be balanced with the processes that establish long-term immunity such as affinity maturation, formation of memory B and T cells, and differentiation of long-lived bone marrow plasma cells.
Much remains unknown about the genetic and systems-level decision-making that guides these short- and long-term processes. Without that understanding there are major gaps in strategies to establish and improve vaccines against many NIH Priority Pathogens, and to interpret patterns of human genetic, cellular, and serological variation to predict the efficacy and longevity of immunity in clinical settings and in the field.
This Project, in synergy with other projects of this Program, will expand the community resource of mouse mutations and datasets that experimentally connect discrete genes with the cells and systems governing short- and long-term adaptive immunity. In addition, the project will focus on a subset of novel gene mutations affecting memory versus effector decisions or antibody affinity maturation in the context of immunity to NIH Priority Pathogens, and determine the cellular and molecular systems controlled by these genes.
Aim 1. Using Systems Guided Forward Genetics, identify mouse strains with novel mutations that alter the formation, persistence or quality of memory and effector T and B cells in response to NIAID Priority A and B pathogens.
This Project will screen for adaptive immune defects in the homozygous and heterozygous mutant progeny of ENU mutant C57BL/6 mice. The screened mice will be produced by the Genetics Core in hundreds of parallel pedigrees each segregating unique protein-changing DNA variants identified by exome sequencing of the G1 mice. Microarray profiling, network analysis, and human exome sequence results will flag mutations in candidate novel immune regulator genes, and these will be analyzed for their consequences on adaptive immunity. These protein-changing DNA variants will be examined using a panel of highly validated adaptive immunity screening systems.
Aim 2. Using the strains developed in Aim 1, define the cellular and molecular systems that are perturbed by selected mutations, and establish how these systems govern the formation of memory and effector T and B cells in response to Category A and B pathogens.
Strains with novel humoral or T cell immune defects revealed in Aim 1 will be propagated and analysed to establish the cell types, stages of development, and molecular pathways or networks directly perturbed by each mutation. Systems profiling of the role of each new gene will be achieved by mixed bone marrow chimera analysis, adoptive transfer of antigen specific B or T cells from transgenic mice, RNA-seq analysis, SRM proteomic analysis, and CyTOF analysis, in conjunction with the Cores.
Aim 3. Using the findings in Aims 1 and 2, validate the activity of these genes and systems in human T and B cells.
The role of novel regulators of T or B cells will be validated in human cells using RNA-knockdown studies in the Human Correlation Core. In parallel, humans with mutations in the same genes will be identified and analyzed side-by-side with the ENU mouse mutants through an international collaborative network.